Running Gaussian on the ORC Clusters
Gaussian16 is available on the ORC clusters. Due to licensing restrictions, only the ORC users in the Gaussian user group are able to use it.
Getting Added to the Gaussian User group
All users on the ORC clusters who want to use Gaussian16 need to sign a user agreement form as per the University Counsel office. Contact the ORC through orchelp@gmu.edu to get a copy of the Gaussian License T&C. You will also be sent the User Agreement to sign and will be added to the Gaussian User Group after the ORC gets back the signed form.
To check the installed versions:
module avail gaussian
------------------------------------------------------------------------------------------------
gaussian:
------------------------------------------------------------------------------------------------
Versions:
gaussian/09
gaussian/16-c01
gaussian/16-c02
------------------------------------------------------------------------------------------------
For detailed information about a specific "gaussian" package (including how to load the modules) use the module's full name.
Note that names that have a trailing (E) are extensions provided by other modules.
For example:
$ module spider gaussian/16-c02
------------------------------------------------------------------------------------------------
and add it to your environment with:
module load gaussian/16-c02
Running Gaussian on CPUs with Slurm
To set up a gaussian run on the cluster, create a directory and add input file and slurm script to it:
mkdir g16-test-cpu
cd g16-test-cpu
./g16-test-cpu
├── [g16-test-cpu.com](g16_files/g16-test-cpu.com)
└── [g16-test-cpu.slurm](g16_files/g16-test-cpu.slurm)
0 directories, 2 files
Example input file g16-test-cpu.com
The example input file show below can be downloaded to your directory from the terminal with
wget http://wiki.orc.gmu.edu/mkdocs/g16_files/g16-test-cpu.com
%nproc=24
%mem=80gb
%chk=jneutral-opt.chk
#T b3lyp/6-31+G* integral=UltraFine opt(maxstep=10,maxcycles=200)
4-BMC-optimization
0 1
O 0.00000 0.00000 0.00000
C 0.00000 0.00000 3.50600
C 0.00000 0.97000 2.28000
C 1.45100 -0.52200 3.25200
C 1.30800 1.79400 2.51700
C 2.28200 0.79500 3.22500
C 1.35400 -0.96600 1.80900
C 0.38700 -0.00800 1.16800
C -0.16600 0.70700 4.85600
C -1.05600 -1.10900 3.39800
C -1.24100 1.79300 1.99500
H 1.79700 -1.29000 3.96000
H 1.08000 2.66500 3.15200
H 1.71000 2.17800 1.56600
H 3.22200 0.66500 2.66700
H 2.53700 1.12700 4.24400
H 0.51000 1.56400 4.98600
H 0.02600 -0.00500 5.67600
H -1.20100 1.07300 4.96800
H -0.91900 -1.84200 4.21200
H -1.00000 -1.65300 2.44100
H -2.06900 -0.68600 3.49300
H -1.09900 2.38900 1.07900
H -1.45400 2.48200 2.82900
H -2.11800 1.14600 1.83500
C 1.93800 -1.98700 1.12200
H 2.25200 -2.85000 1.72600
C 2.22300 -2.03300 -0.31600
C 1.45800 -1.31800 -1.27000
C 3.28700 -2.84400 -0.78300
C 1.77600 -1.38900 -2.63700
H 0.59000 -0.73900 -0.94700
C 3.60200 -2.90100 -2.14900
H 3.87200 -3.42400 -0.06100
C 2.85600 -2.17000 -3.10000
H 1.16500 -0.83800 -3.36000
H 4.43500 -3.52900 -2.48500
C 3.20900 -2.23400 -4.56900
H 3.26000 -3.27800 -4.91800
H 2.46300 -1.70300 -5.18000
H 4.19300 -1.77200 -4.75600
Example Slurm script g16-test-cpu.slurm
This example slurm script can be downloaded to your directory from the terminal with
wget http://wiki.orc.gmu.edu/mkdocs/g16_files/g16-test-cpu.slurm
#!/bin/bash
#SBATCH -p normal
#SBATCH -J g16-test-cpu
#SBATCH -o g16-test-cpu-%j-%N.out
#SBATCH -e g16-test-cpu-%j-%N.err
#SBATCH --nodes=1
#SBATCH --ntasks-per-node=24
#SBATCH --mem-per-cpu=3500MB
#SBATCH --export=ALL
#SBATCH -t 00:30:00
set echo
umask 0027
echo $SLURM_SUBMIT_DIR
module load gaussian/16-c01
module list
# default scratch location is /scratch/$USER/gaussian. Users can change it using
# export GAUSS_SCRDIR=<NEW_SCRATCH_LOCATION>
which g16
g16 < g16-test-cpu.com > g16-test-cpu.log
Submitting the job
Once you have the files, submit the job to the cluster with:
sbatch g16-test-cpu.slurm
./g16-test-cpu
├── g16-test-cpu-66631-hop049.err
├── g16-test-cpu-66631-hop049.out
├── g16-test-cpu.com
├── g16-test-cpu.log
├── g16-test-cpu.slurm
└── jneutral-opt.chk
To monitor the job and check on its status, you can use the slurm command
sacct -X --format=jobid,jobname,partition,state
JobID JobName Partition State
------------ ---------- ---------- ----------
66631 g16-test-+ normal COMPLETED
Running Gaussian on a GPU with Slurm
The Gaussian module installed on Hopper can be run on one of the A100.80gb gpu nodes. For more information about the gpus available on the cluster and running gpu jobs, please refer to this page.
Setting the gpucpu options in the Gaussian input file
To set the gpucpu options in the Gaussian input file, you need to add the following line to the input file:
%gpucpu=cpu_list=gpu_list
where - cpu_list is a comma-separated list of CPU numbers that will be used to control the GPUs, and - gpu_list is a comma-separated list of GPU numbers that will be used for the calculation.
For example, if you want to use two GPUs and the CPU numbers are 0 and 1 and the GPU numbers are 0 and 1, you would add the following line to the Gaussian input file:
%gpucpu=0,1=0,1
To determine the control cpus for any gpus on the cluster, you need to be logged on to the gpu and run:
nvidia-smi topo -m
For the Hopper A100.80GB AMD nodes, this should produce output similar to:
GPU0 GPU1 GPU2 GPU3 mlx5_0 mlx5_1 mlx5_2 mlx5_3 mlx5_4 CPU Affinity NUMA Affinity
GPU0 X NV4 NV4 NV4 SYS SYS SYS SYS SYS 24-31 3
GPU1 NV4 X NV4 NV4 SYS SYS SYS SYS SYS 8-15 1
GPU2 NV4 NV4 X NV4 SYS SYS SYS SYS SYS 56-63 7
GPU3 NV4 NV4 NV4 X SYS SYS SYS SYS SYS 40-47 5
mlx5_0 SYS SYS SYS SYS X PIX SYS SYS SYS
mlx5_1 SYS SYS SYS SYS PIX X SYS SYS SYS
mlx5_2 SYS SYS SYS SYS SYS SYS X SYS SYS
mlx5_3 SYS SYS SYS SYS SYS SYS SYS X PIX
mlx5_4 SYS SYS SYS SYS SYS SYS SYS PIX X
Legend:
X = Self
SYS = Connection traversing PCIe as well as the SMP interconnect between NUMA nodes (e.g., QPI/UPI)
NODE = Connection traversing PCIe as well as the interconnect between PCIe Host Bridges within a NUMA node
PHB = Connection traversing PCIe as well as a PCIe Host Bridge (typically the CPU)
PXB = Connection traversing multiple PCIe bridges (without traversing the PCIe Host Bridge)
PIX = Connection traversing at most a single PCIe bridge
NV# = Connection traversing a bonded set of # NVLinks
%GPUCPU=0,1,2,3=24,8,56,40
Tips for setting the gpucpu options in the Gaussian input file:
- There are 4 GPUs on each gpu node, and Gaussian will only run on a single gpu node. Make sure that the number of GPUs that you specify on the %gpucpu line is less than or equal to the number of GPUs that are available on your system.
- Make sure that the CPU numbers that you specify on the %gpucpu line are available on your system.
- Make sure that the GPU numbers that you specify on the %gpucpu line are available on your system.
Depending on the size of the system being calculated, you can set your gpucpu option for a single gpu with:
%GPUCPU=0=24
or any of the other gpus, 1,2,3 as long as they are matched with the correct control cpu. Increase the number of gpus (to a maximum of 4) as needed.
Once you have set the gpucpu options in the Gaussian input file, you can run Gaussian as usual.
Example input file for gpu run g16-test-gpu.com
The example input file show below can be downloaded to your directory from the terminal with
wget http://wiki.orc.gmu.edu/mkdocs/g16_files/g16-test-gpu.com
Create a Slurm job submission script for a GPU run
The following is an example of a Slurm job submission script g16-test-gpu.slurm that can be used to run Gaussian on a GPU:
#!/bin/bash
#SBATCH --job-name g16-test-gpu
#SBATCH -p gpuq
#SBATCH -q gpu
#SBATCH --gres=gpu:A100.80gb:4
#SBATCH --mem=350GB
#SBATCH --nodes=1
#SBATCH --ntasks-per-node=64
#SBATCH -o PP_SPIN_1_%j.out # output file name
#SBATCH -e PP_SPIN_1_%j.err # err file name
#SBATCH --export=ALL
#SBATCH -t 00-1:00:00 # set to 1hr; please choose carefully
set echo
umask 0027
echo $SLURM_SUBMIT_DIR
module load gnu10 openmpi
module load gaussian/16-c02 # load relevant modules
module list
# default scratch location is /scratch/$USER/gaussian. Users can change it using
# export GAUSS_SCRDIR=<NEW_SCRATCH_LOCATION>
which g16
g16 < PP_SPIN_1.com > PP_SPIN_1_gpu.log # activate the virtual environment
This script will ask for 4 gpus and 64 cpus on one of the A100.80gb nodes. To download it, use:
wget http://wiki.orc.gmu.edu/mkdocs/g16_files/g16-test-gpu.slurm
Once you're ready, submit the Slurm job submission script to the Slurm scheduler with:
sbatch g16-test-gpu.slurm
Wait for the Slurm job to finish.
Check the output of the Slurm job to see if the Gaussian calculation was successful.
Using GaussView on the Open OnDemand Server
*** NOTE: *** ** Tutorials on using GaussView for different processes can be found on these Gaussian Pages **
To use GaussView on Hopper, you need to start an Open OnDemand (OOD)session. In your browser, go to ondemand.orc.gmu.edu. You will need to authenticate with your Mason credentials before you can access the dashboard.
- Once you authenticate, you should be directed to the OOD dashboard.
- You'll need to click on the "Interactive Apps icon to see the apps that are available.
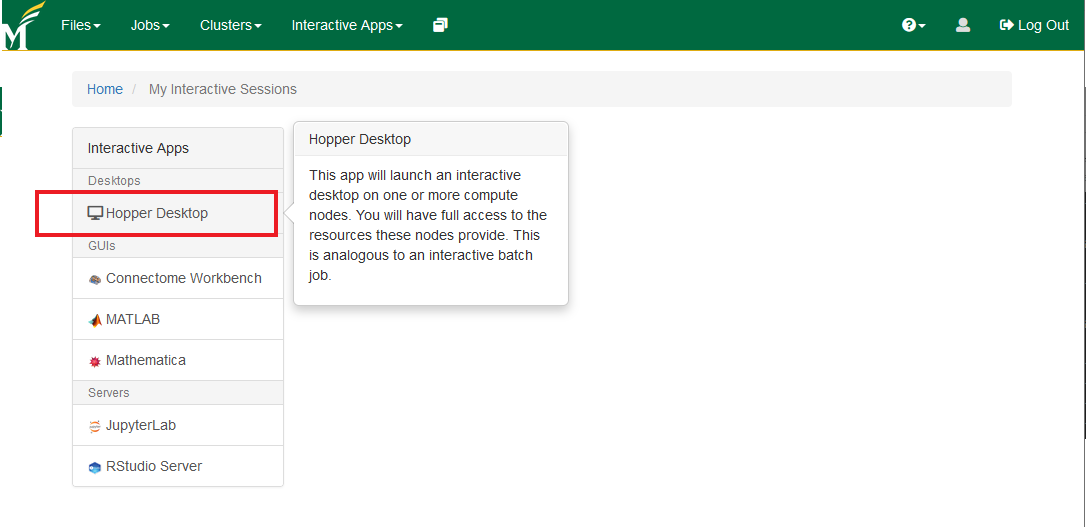
- Select the Hopper Desktop and set your configuration (number of hours upto 12 and number of cores upto 12). You'll see the launch option once the resources become available.
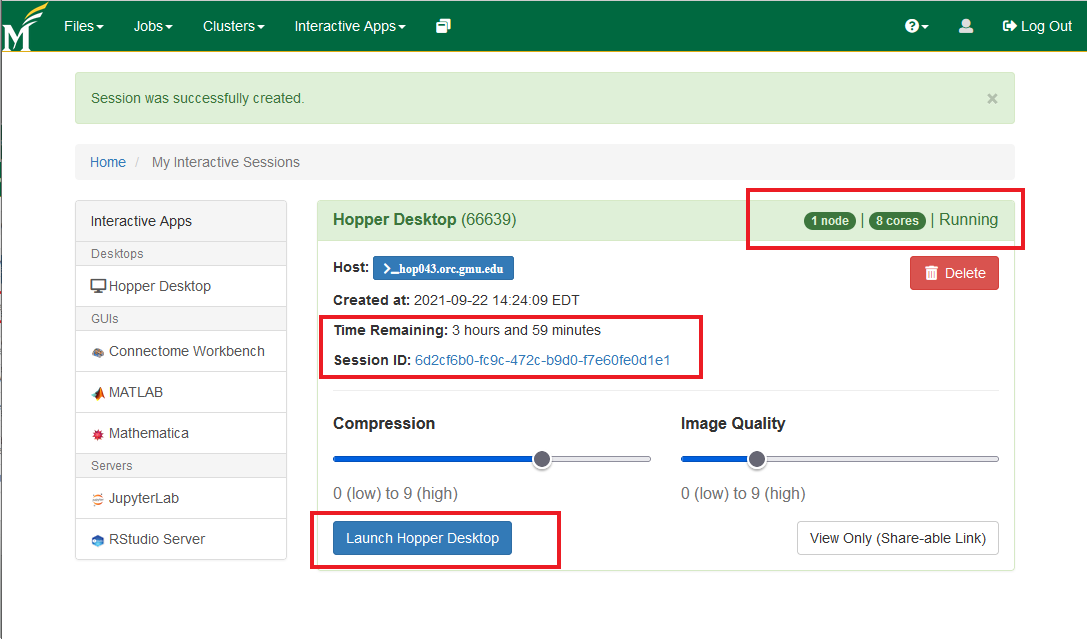
- After launching the session, you'll get connected to a node on Hopper with a desktop environment from where you can now start the terminal (using the highlighted 'terminal emulator' icon)
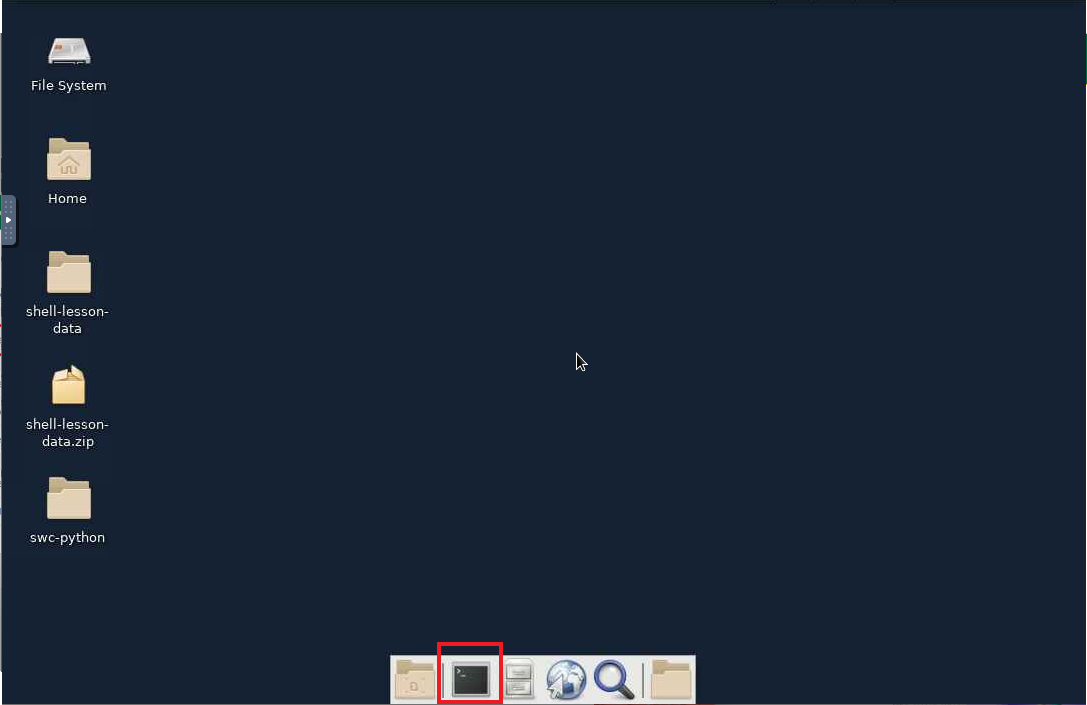
- From the launched shell session, load the gaussian module with
module load gaussian
gv
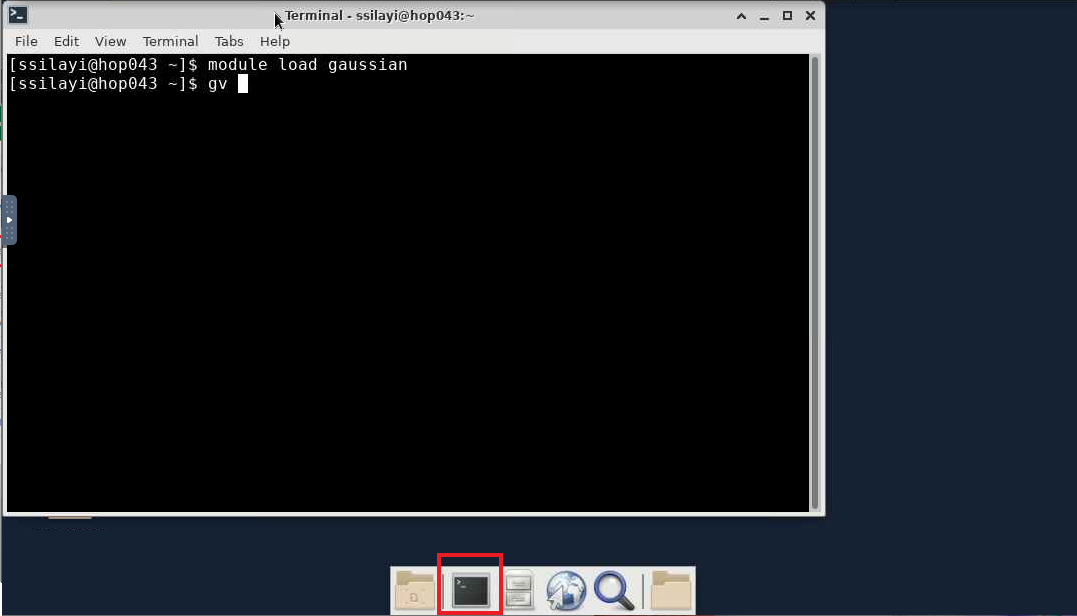
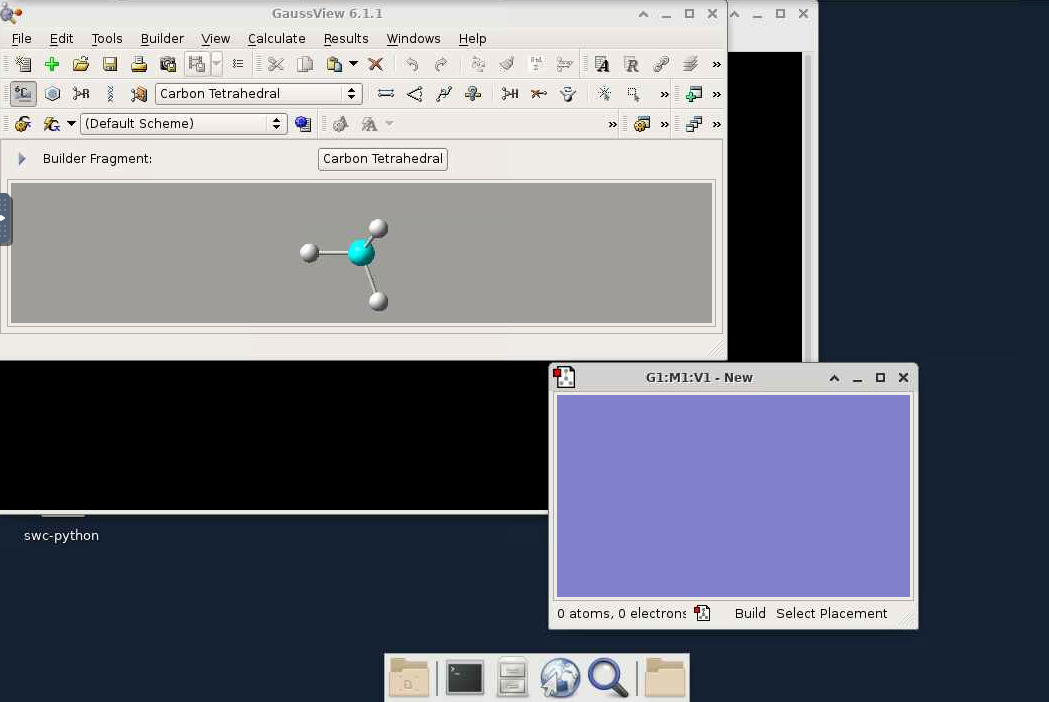
- The GaussView window should now appear
Useful resources for Troubleshooting Gaussian Errors
A lot of the common Gaussian Errors and possible solutions have been curated on this page here: Gaussian Error Messages